%matplotlib inline
Landmark-based multi-sample single-cell data analysis
# Table of contents * Read Data * Generate Sample Embeddings * Samaple-based AnalysisGet started
To run this notebook, you have to download the
preterm_unstim.h5addata from this link, and put it into thedatafolder under the project root.Install our
sclkmepackage and other dependencies using thepipcommand:pip install sclkme phate statannotations
If there are any questions or problems, please push an issue under the git repo at https://github.com/CompCy-lab/scLKME.
# Uncomment and run this cell if you're on Colab or Kaggle
# !pip install sclkme phate statannotations
# load the packages needed
%reload_ext autoreload
# load the dependencies
import sclkme # load the sclkme package
import scanpy as sc
import anndata as ad
import os.path as osp
import pandas as pd
import matplotlib.pyplot as plt
import matplotlib.ticker as ticker
import matplotlib as mpl
mpl.rcParams["figure.facecolor"] = 'white'
import seaborn as sns
from pathlib import Path
from matplotlib.pyplot import rc_context
sc.settings.verbosity = 1
sc.settings.set_figure_params(dpi=80, facecolor='white')
sc.logging.print_header()
scanpy==1.9.3 anndata==0.9.2 umap==0.5.4 numpy==1.23.5 scipy==1.10.1 pandas==1.5.3 scikit-learn==1.3.2 statsmodels==0.14.0 python-igraph==0.10.8 pynndescent==0.5.10
Read the data
data_root = osp.join(osp.dirname(osp.abspath("__file__")), "../../data")
adata_path = osp.join(Path(data_root).resolve(), "preterm_unstim.h5ad")
# read the .h5ad data
adata = sc.read_h5ad(adata_path)
# show the available fields in the data
print(adata)
AnnData object with n_obs × n_vars = 400000 × 38
obs: 'Sample_ID', 'Plate_ID', 'Stim', 'FCS_File', 'Class', 'Category', 'Gestational.Age', 'X_64_sketch', 'X_128_sketch', 'X_256_sketch', 'X_512_sketch'
uns: 'X_128_sketch', 'X_256_sketch', 'X_512_sketch', 'X_64_sketch', 'kme', 'neighbors', 'sampleXmeta', 'umap'
obsm: 'X_kme', 'X_umap'
obsp: 'connectivities', 'distances'
Generate sample-based embeddings
The landmark matrix X_anchor is an n_landmark by d matrix, where the rows are landmark cells and the columns are the embedding features, which can be raw gene/protein features or extracted features, like PCA.
# cell sketching with n_landmark=512 landmarks
sclkme.tl.sketch(adata, n_sketch=512, use_rep="X", key_added="X_512", method="kernel_herding")
adata_sketch = adata[adata.obs['X_512_sketch']]
# get the precomputed anchor cells
X_anchor = adata_sketch.X.copy()
print(X_anchor)
[[0.06662848 4.0252423 3.2229903 ... 1.0307382 0.3225428 0.1303769 ]
[1.4203637 4.2610917 2.4013813 ... 0. 0. 0. ]
[0.6415793 4.6462755 3.3441646 ... 0.5812172 1.8201672 0. ]
...
[0.40527838 4.117027 3.5839536 ... 0.65212226 0.49960423 0. ]
[0. 4.126464 4.0653095 ... 0. 0. 0. ]
[0.8006469 4.363835 3.8853781 ... 1.1542339 3.6173465 1.370174 ]]
Run the kernel mean embedding using the api provided by the sclkme package.
This api is pretty much similar to standard interface in the scanpy ecosystem. For the parameters,
partition_key: the column name of sample ids in theadata.obsdataframe.X_anchor: the landmakr matrixuse_rep: the indicated representation to be used. Note: the representation should have identical dimensions as the landmark matrix.
By default, the output sample embeddings are stored in adata.uns['kme']['{partition_key}_kme'].
# run kernel mean embedding
sclkme.tl.kernel_mean_embedding(adata, partition_key="Sample_ID", X_anchor=X_anchor, use_rep='X')
# show sample embeddings
# pd.set_option('display.max_columns', 6)
adata.uns['kme']['Sample_ID_kme'].head()
| X_kme_0 | X_kme_1 | X_kme_2 | X_kme_3 | X_kme_4 | X_kme_5 | X_kme_6 | X_kme_7 | X_kme_8 | X_kme_9 | ... | X_kme_502 | X_kme_503 | X_kme_504 | X_kme_505 | X_kme_506 | X_kme_507 | X_kme_508 | X_kme_509 | X_kme_510 | X_kme_511 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample_ID | |||||||||||||||||||||
| CB10T | 0.568137 | 0.516859 | 0.647542 | 0.362659 | 0.454447 | 0.589324 | 0.600191 | 0.483299 | 0.593141 | 0.372919 | ... | 0.442153 | 0.381815 | 0.524996 | 0.485006 | 0.636462 | 0.608896 | 0.575762 | 0.410888 | 0.420804 | 0.497886 |
| CB11-AP | 0.509170 | 0.522249 | 0.514000 | 0.407897 | 0.471045 | 0.468700 | 0.494871 | 0.397684 | 0.471713 | 0.352793 | ... | 0.489170 | 0.431116 | 0.510078 | 0.458123 | 0.627160 | 0.547009 | 0.525865 | 0.480013 | 0.464187 | 0.459410 |
| CB11-BP | 0.510515 | 0.504690 | 0.529104 | 0.450097 | 0.510501 | 0.471506 | 0.499843 | 0.442229 | 0.475539 | 0.339085 | ... | 0.485501 | 0.461806 | 0.523375 | 0.478437 | 0.643484 | 0.571422 | 0.552002 | 0.465727 | 0.453075 | 0.474139 |
| CB12P | 0.452424 | 0.521209 | 0.406091 | 0.513055 | 0.545144 | 0.346239 | 0.387355 | 0.392161 | 0.352428 | 0.385342 | ... | 0.520035 | 0.469245 | 0.400201 | 0.337749 | 0.529777 | 0.432825 | 0.409252 | 0.503371 | 0.491035 | 0.336433 |
| CB14P | 0.507396 | 0.620306 | 0.459013 | 0.399986 | 0.441434 | 0.413071 | 0.416108 | 0.335017 | 0.411108 | 0.540626 | ... | 0.599916 | 0.393721 | 0.405664 | 0.324018 | 0.508857 | 0.435034 | 0.397705 | 0.593167 | 0.599104 | 0.334227 |
5 rows × 512 columns
Sample-based data analysis
# acquire sample-level meta data
sample_obs = adata.obs[["Sample_ID", "Class", "Category", "Gestational.Age"]].groupby(by="Sample_ID", sort=False).agg(lambda x: x[0])
adata_sample = ad.AnnData(adata.uns['kme']['Sample_ID_kme'], obs=sample_obs)
/Users/haidyi/Documents/proj/scLKME/.venv/lib/python3.10/site-packages/anndata/_core/anndata.py:121: ImplicitModificationWarning: Transforming to str index.
warnings.warn("Transforming to str index.", ImplicitModificationWarning)
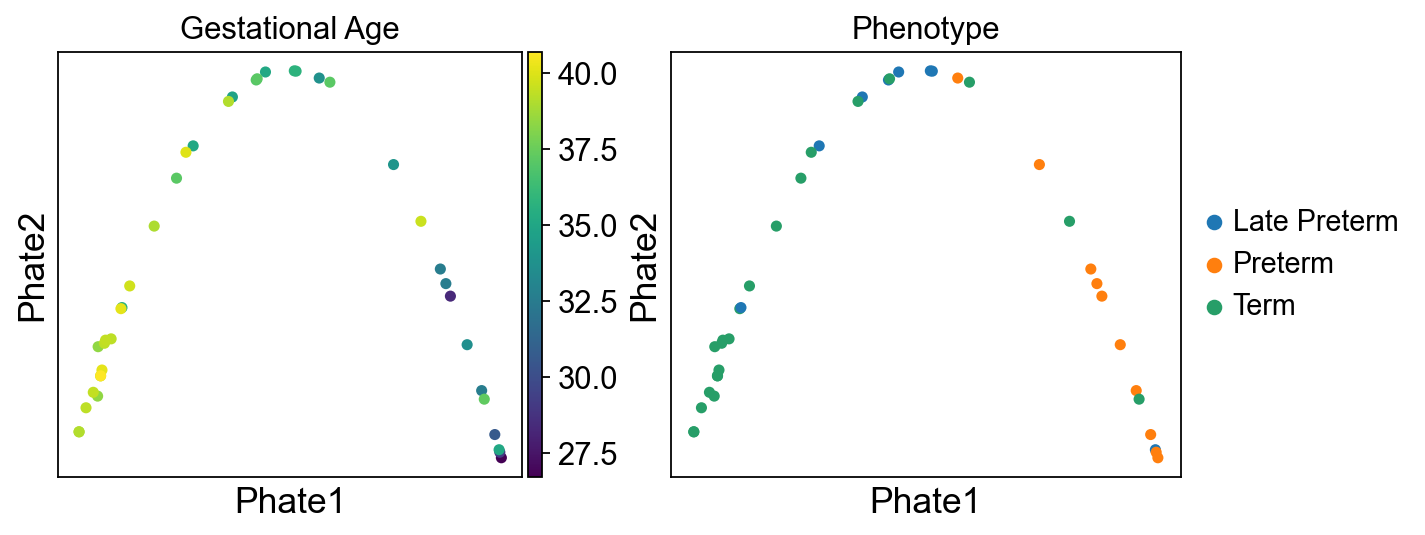
Visualize sample embeddings using PHATE
sc.external.tl.phate(adata_sample, n_pca=10, k=9, t="auto",
random_state=0, mds_solver="smacof", verbose=True, copy=False)
fig, axes = plt.subplots(1, 2, figsize=(9, 3.5))
sc.external.pl.phate(adata_sample, color=['Gestational.Age'], title="Gestational Age", color_map="viridis", size=100, ax=axes[0], show=False)
sc.external.pl.phate(adata_sample, color=['Category'], title="Phenotype", size=100, ax=axes[1], show=False)
for ax in axes:
ax.set_xlabel("Phate1", fontsize=16)
ax.set_ylabel("Phate2", fontsize=16)
plt.show()
/Users/haidyi/Documents/proj/scLKME/.venv/lib/python3.10/site-packages/pygsp/filters/simpletight.py:79: SyntaxWarning: "is" with a literal. Did you mean "=="?
if kerneltype is 'sf':
/Users/haidyi/Documents/proj/scLKME/.venv/lib/python3.10/site-packages/pygsp/filters/simpletight.py:82: SyntaxWarning: "is" with a literal. Did you mean "=="?
elif kerneltype is 'wavelet':
Calculating PHATE...
Running PHATE on 40 observations and 512 variables.
Calculating graph and diffusion operator...
Calculating PCA...
Calculated PCA in 0.01 seconds.
Calculating KNN search...
Calculating affinities...
Calculated graph and diffusion operator in 0.03 seconds.
Calculating optimal t...
Automatically selected t = 23
Calculated optimal t in 0.02 seconds.
Calculating diffusion potential...
Calculating metric MDS...
Calculated metric MDS in 0.01 seconds.
Calculated PHATE in 0.08 seconds.
/Users/haidyi/Documents/proj/scLKME/.venv/lib/python3.10/site-packages/phate/phate.py:186: FutureWarning: k is deprecated. Please use knn in future.
warnings.warn("k is deprecated. Please use knn in future.", FutureWarning)
/Users/haidyi/Documents/proj/scLKME/.venv/lib/python3.10/site-packages/phate/phate.py:190: FutureWarning: a is deprecated. Please use decay in future.
warnings.warn("a is deprecated. Please use decay in future.", FutureWarning)
/Users/haidyi/Documents/proj/scLKME/.venv/lib/python3.10/site-packages/sklearn/manifold/_mds.py:298: FutureWarning: The default value of `normalized_stress` will change to `'auto'` in version 1.4. To suppress this warning, manually set the value of `normalized_stress`.
warnings.warn(
/Users/haidyi/Documents/proj/scLKME/.venv/lib/python3.10/site-packages/scanpy/plotting/_tools/scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
Visualize sample embeddings using diffusion map
sc.pp.neighbors(adata_sample, n_neighbors=8, use_rep="X", key_added="diffmap_nn")
sc.tl.diffmap(adata_sample, n_comps=3, neighbors_key="diffmap_nn")
sc.pl.diffmap(adata_sample, color=["Gestational.Age", "Category"], size=100,
palette=sc.pl.palettes.vega_20_scanpy, color_map="viridis", components=['2,3'])
/Users/haidyi/Documents/proj/scLKME/.venv/lib/python3.10/site-packages/scanpy/plotting/_tools/scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(

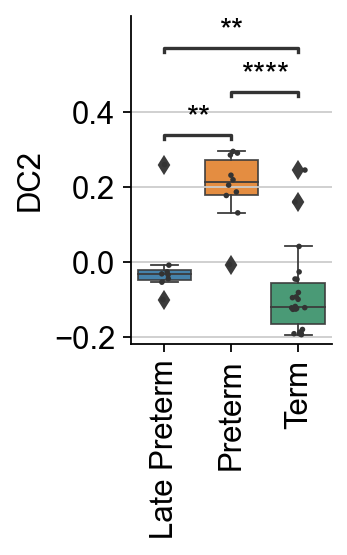
T-test on the coordinates of diffusion map
from statannotations.Annotator import Annotator
diffmap_df = sc.get.obs_df(adata_sample, keys=['Category', 'Gestational.Age'], obsm_keys=[('X_diffmap', 1)])
g = sns.catplot(x="Category", y="X_diffmap-1", data=diffmap_df, kind="box",
legend=False, linewidth=0.8, boxprops=dict(alpha=.9), height=3, aspect=0.8, sharey=False)
axes = g.axes.flatten()
for ax, y in zip(g.axes.flatten(), ["X_diffmap-1"]):
sns.stripplot(x="Category", y=y, data=diffmap_df, color=".2", size=2.5, jitter=True, ax=ax)
annot = Annotator(ax, [("Term", "Late Preterm"), ("Late Preterm", "Preterm"), ("Term", "Preterm")],
data=diffmap_df, x="Category", y=y, order=["Late Preterm", "Preterm", "Term"])
annot.configure(test='Mann-Whitney', text_format='star', loc='inside', verbose=2)
annot.apply_test()
_, test_results = annot.annotate()
for ax in axes:
ax.set_title(None)
ax.set_xticklabels(labels=ax.get_xticklabels(), rotation=90)
axes[0].set_ylabel("DC2")
axes[0].set_yticks([-0.2, 0.0, 0.2, 0.4])
g.set_xlabels("")
plt.show()
p-value annotation legend:
ns: 5.00e-02 < p <= 1.00e+00
*: 1.00e-02 < p <= 5.00e-02
**: 1.00e-03 < p <= 1.00e-02
***: 1.00e-04 < p <= 1.00e-03
****: p <= 1.00e-04
Late Preterm vs. Preterm: Mann-Whitney-Wilcoxon test two-sided, P_val:2.057e-03 U_stat=7.000e+00
Preterm vs. Term: Mann-Whitney-Wilcoxon test two-sided, P_val:5.227e-05 U_stat=2.100e+02
Late Preterm vs. Term: Mann-Whitney-Wilcoxon test two-sided, P_val:9.928e-03 U_stat=1.420e+02